Welcome to the NoMoRe server!
What is Normal Mode Refinement?
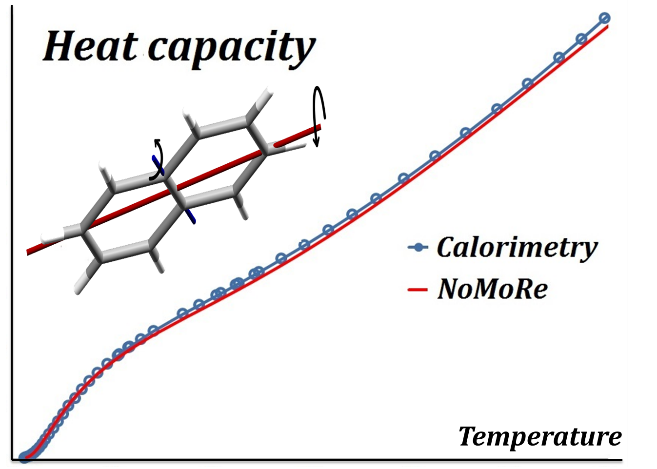
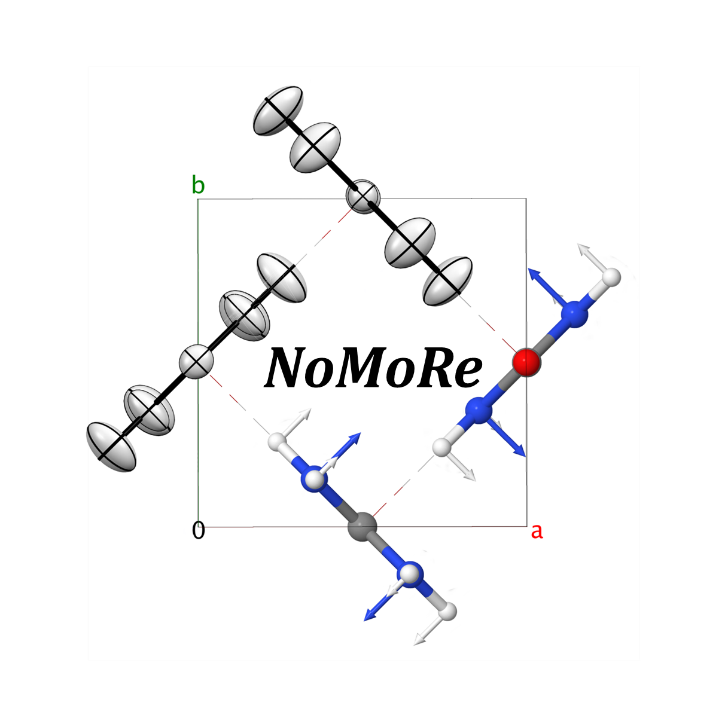
Normal Mode Refinement (NoMoRe) is a completely new modelling approach, which enables the refinement of frequencies of normal modes evaluated using ab-initio periodic computations against single crystal diffraction data. In this approach instead of refining anisotropic displacement parameters (ADPs) as in routine X-ray refinement, we are refining only few frequencies related to low-frequency modes, which corresponds to the external vibrations of molecule.
Schematic workflow of our method is presented in Scheme 1. The frequencies, which are obtained after the refinement, enable calculation of thermodynamic properties.

Scheme 1. Schematic workflow with Normal Mode Refinement (NoMoRe).
What are advantages of using NoMoRe
-
With fewer parameters we can obtain results of very similar quality as the standard ADP refinements
-
Obtained frequencies can be used for evaluation of thermodynamic properties (heat capacity, vibrational entropy and enthalpy contributions to free energy)
-
H atom ADPs in very good agreement with neutron value
For more details related to NoMoRe see:
-
Hoser A. A., Madsen A. Ø., Acta Cryst A 2016, A72, 206-214
-
Hoser A. A., Madsen A. Ø., Acta Cryst A 2017, A73, 102-114
-
Kofoed, P. M., Hoser, A. A., Diness, F., Capelli, S. C., Madsen, A. Ø., IUCrJ, 2019
How to run NoMoRe?
Because calculations can be time-consuming we decided to create an accounts for users. After calculations, the results will be available from the user account in the dashboard section.
Plese create an account and sign in with Login/Sign up section on the top right corner of screen.
After login in the menu two new options will appear
- Dashboard - hence, you will be able to download the results file
- Add - form for ordering new calculations
In order to start NoMoRe three files are needed:
-
Results of periodic ab-initio Gamma point frequency calculations - from CRYSTAL09 or CRYSTAL14 or CRYSTAL17 – input for such calculations can be generated by cif2crystal program (http://shade.ki.ku.dk/docs/cif2crystal.html)
-
Structure factors files in shelx format (hkl file)
-
Input file for shelx (.ins file)
Here are examples of input files
In addition, the following fields are available in the form. (Those with a default value are required.)
- name and descroption - for identification purposes
-
others parameters like: (temperature, number of refined frequencies)
Please find manual here
Important
Please notice that this server is in development. Your comments and bug reports will be greatly appreciated and we will try to respond quickly with an updated version. We recommend that you critically inspect the resulting ADPs using a visualization program, e.g. Ortep, Platon, Mercury or Peanut.
Bug reports, requests for improvements and all other sorts of comments are greatly appreciated and should be sent to nomore_help@chem.uw.edu.pl
Contact
Anna A. Hoser
University of Warsaw,
Chemistry Department
e-mail: a.hoser@uw.edu.pl
https://crystal.chem.uw.edu.pl/people/wozniaks-group/staff/hoser/
Anders Ø. Madsen
University of Copenhagen
Department of Pharmacy
e-mail: a.madsen@sund.ku.dk
Founding
Foundation for Polish Science
Villum Foundation